Free-energy perturbation (FEP) is an alchemical method based on statistical mechanics that is used in computational chemistry for computing free-energy differences from molecular dynamics or Metropolis Monte Carlo simulations. It is widely used to compute the free energy difference between two states, state A and state B.
The FEP method was introduced by Robert W. Zwanzig in 1954.1 According to the free-energy perturbation method, the free-energy difference for going from state A to state B is obtained from the following equation, known as the Zwanzig equation:
where T is the temperature, kB is the Boltzmann constant, and the angular brackets denote an average over a simulation run for state A. In practice, one runs a normal simulation for state A, but each time a new configuration is accepted, the energy for state B is also computed. The difference between states A and B may be in the atom types involved, in which case the ΔF obtained is for "mutating" one molecule onto another, or it may be a difference of geometry, in which case one obtains a free-energy map along one or more reaction coordinates. This free-energy map is also known as a potential of mean force (PMF).
Free-energy perturbation calculations only converge properly when the difference between the two states is small enough (the phase space of the two states overlap). In practice, the perturbation is divided into a series of smaller, discrete intermediate FEP windows defined by the coupling parameter . A value of = 0 represent 100% state A, and = 1 represent 100% state B. In any particular window, we have the effective potential energy function , which is the linear combination of the initial potential A and final B. Stepwise varying from 0 to 1 creates an "alchemical" pathway of transformation between state A and B through non-physical intermediate states.2
Since there is no need for constant communication between the simulation for one window and the next, the process can be trivially parallelized by running each window on a different CPU, in what is known as an "embarrassingly parallel" setup.
Application
FEP calculations have been used for studying host–guest binding energetics, pKa predictions, solvent effects on reactions, and enzymatic reactions. Other applications are the virtual screening of ligands in drug discovery, in silico mutagenesis studies34 and antibody affinity maturation.5 For the study of reactions it is often necessary to involve a quantum-mechanical (QM) representation of the reaction center because the molecular mechanics (MM) force fields used for FEP simulations cannot handle breaking bonds. A hybrid method that has the advantages of both QM and MM calculations is called QM/MM.
Umbrella sampling is another free-energy calculation technique that is typically used for calculating the free-energy change associated with a change in "position" coordinates as opposed to "chemical" coordinates, although umbrella sampling can also be used for a chemical transformation when the "chemical" coordinate is treated as a dynamic variable (as in the case of the Lambda dynamics approach of Kong and Brooks). An alternative to free-energy perturbation for computing potentials of mean force in chemical space is thermodynamic integration. Another alternative, which is probably more efficient, is the Bennett acceptance ratio method. Adaptations to FEP exist which attempt to apportion free-energy changes to subsections of the chemical structure.6
Thermodynamic Cycle
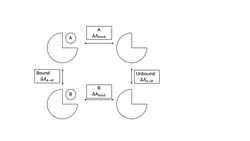
Since free energy is a state function, the total free energy change between two states are independent of the path taken. Using FEP calculations and closed thermodynamic cycles, it is possible to calculate the relative change in free energy, for example comparing two ligands A and B binding to a target substrate. From this we can calculate the change in binding free energy as 7
Software
FEP methods have been implemented in several molecular dynamics software packages. Below is a short list of some of the most common programs:
- Flare FEP8
- FEP+9
- AMBER10
- BOSS
- CHARMM
- Desmond
- GROMACS
- OpenMM
- MacroModel
- MOLARIS11
- NAMD
- Tinker
- Q
- QUELO12
References
References
- Zwanzig, R. W. J. Chem. Phys. 1954, 22, 1420–1426. doi:10.1063/1.1740409.
- Jespers, Willem; Isaksen, Geir V.; Andberg, Tor A.H.; Vasile, Silvana; van Veen, Amber; Åqvist, Johan; Brandsdal, Bjørn Olav; Gutiérrez-de-Terán, Hugo (2019-10-08). "QresFEP: An Automated Protocol for Free Energy Calculations of Protein Mutations in Q". Journal of Chemical Theory and Computation. 15 (10): 5461–5473. Bibcode:2019JCTC...15.5461J. doi:10.1021/acs.jctc.9b00538. hdl:10037/18010. ISSN 1549-9618. PMID 31436990.
- Sampson, Jared M.; Cannon, Daniel A.; Duan, Jianxin; Epstein, Jordan C. K.; Sergeeva, Alina P.; Katsamba, Phinikoula S.; Mannepalli, Seetha M.; Bahna, Fabiana A.; Adihou, Hélène (2024). "Robust Prediction of Relative Binding Energies for Protein–Protein Complex Mutations Using Free Energy Perturbation Calculations". Journal of Molecular Biology. 436 (16) 168640. bioRxiv 10.1101/2024.04.22.590325. doi:10.1016/j.jmb.2024.168640. PMC 11339910. PMID 38844044.
- Jespers, Willem; Åqvist, Johan; Gutiérrez-de-Terán, Hugo (2021), "Free Energy Calculations for Protein–Ligand Binding Prediction", in Ballante, Flavio (ed.), Protein-Ligand Interactions and Drug Design, Methods in Molecular Biology, vol. 2266, New York, NY: Springer US, pp. 203–226, doi:10.1007/978-1-0716-1209-5_12, ISBN 978-1-0716-1209-5, PMID 33759129, S2CID 226701336, retrieved 2021-03-30.
- Sampson, Jared M.; Cannon, Daniel A.; Duan, Jianxin; Epstein, Jordan C. K.; Sergeeva, Alina P.; Katsamba, Phinikoula S.; Mannepalli, Seetha M.; Bahna, Fabiana A.; Adihou, Hélène (2024). "Robust Prediction of Relative Binding Energies for Protein–Protein Complex Mutations Using Free Energy Perturbation Calculations". Journal of Molecular Biology. 436 (16) 168640. bioRxiv 10.1101/2024.04.22.590325. doi:10.1016/j.jmb.2024.168640. PMC 11339910. PMID 38844044.
- Irwin, B. W. J., J. Chem. Theory Comput. 2018, 14, 6, 3218–3227. doi:10.1021/acs.jctc.8b00027.
- Jespers, Willem; Esguerra, Mauricio; Åqvist, Johan; Gutiérrez-de-Terán, Hugo (December 2019). "QligFEP: an automated workflow for small molecule free energy calculations in Q". Journal of Cheminformatics. 11 (1) 26. doi:10.1186/s13321-019-0348-5. ISSN 1758-2946. PMC 6444553. PMID 30941533.
- "Flare FEP | Cresset". www.cresset-group.com.
- "FEP+ | Schrödinger". www.schrodinger.com.
- "The Amber Molecular Dynamics Package". ambermd.org.
- "Archived copy". Archived from the original on 2014-12-28. Retrieved 2015-01-18.
{{cite web}}: CS1 maint: archived copy as title (link) - "QSimulate". www.qsimulate.com.