ChIP-exo is a chromatin immunoprecipitation based method for mapping the locations at which a protein of interest (transcription factor) binds to the genome. It is a modification of the ChIP-seq protocol, improving the resolution of binding sites from hundreds of base pairs to almost one base pair. It employs the use of exonucleases to degrade strands of the protein-bound DNA in the 5'-3' direction to within a small number of nucleotides of the protein binding site. The nucleotides of the exonuclease-treated ends are determined using some combination of DNA sequencing, microarrays, and PCR. These sequences are then mapped to the genome to identify the locations on the genome at which the protein binds.
Theory
Chromatin immunoprecipitation (ChIP) techniques have been in use since 19841 to detect protein-DNA interactions. There have been many variations on ChIP to improve the quality of results. One such improvement, ChIP-on-chip (ChIP-chip), combines ChIP with microarray technology. This technique has limited sensitivity and specificity, especially in vivo where microarrays are constrained by thousands of proteins present in the nuclear compartment, resulting in a high rate of false positives.2 Next came ChIP-sequencing (ChIP-seq), which combines ChIP with high-throughput sequencing.3 However, the heterogeneous nature of sheared DNA fragments maps binding sites to within ±300 base pairs, limiting specificity. Secondly, contaminating DNA presents a grave problem since so few genetic loci are cross-linked to the protein of interest, making any non-specific genomic DNA a significant source of background noise.4
To address these problems, Rhee and Pugh revised the classic nuclease protection assay to develop ChIP-exo.5 This new ChIP technique relies on a lambda exonuclease that degrades only, and all, unbound double-stranded DNA in the 5′ to 3′ direction.
Workflow
ChIP
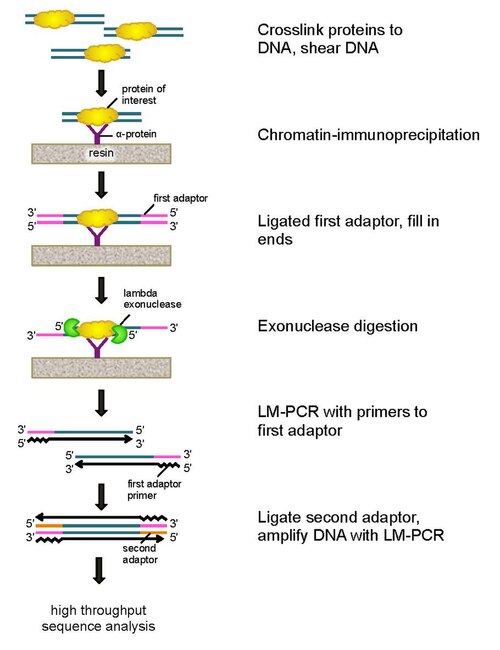
Cells are crosslinked in vivo with formaldehyde to covalently bind proteins to DNA at their natural binding locations across a genome. Cells are then collected, broken open, and the chromatin sheared and solubilized by sonication. An antibody is then used to immunoprecipitate the protein of interest (engineering cells with an epitope tag can be useful for immunoprecipitation), along with the crosslinked DNA. DNA PCR adaptors are then ligated to the ends, which serve as a priming point for second strand DNA synthesis after the exonuclease digestion. Lambda exonuclease then digests double DNA strands from the 5′ end until digestion is blocked at the border of the protein-DNA covalent interaction. Most contaminating DNA is degraded by the addition of a second single-strand specific exonuclease. After the cross-linking is reversed, the primers to the PCR adaptors are extended to form double stranded DNA, and a second adaptor is ligated to 5′ ends to demarcate the precise location of exonuclease digestion cessation. The library is then amplified by PCR, and the products are identified by high throughput sequencing. This method allows for resolution of up to a single base pair for any protein binding site within any genome, which is a much higher resolution than either ChIP-chip or ChIP-seq.
Sequencing
ChIP-exo utilizes short read (e.g. Illumina NGS) sequencing. Sequencing requirements are lower for ChIP-exo than that of other assays like ChIP-seq because the dramatically reduced "shouldering" of a higher resolution assay like ChIP-exo means that the sampling of DNA fragments for constructing the DNA library are better dominated by target-bound sites (this effect can vary across different targets).6
For paired end data from a standard ChIP-exo prep, the 5' end of Read 1 sequenced from the DNA fragments marks the position of the cross-linking site (lambda exonuclease digestion stop site). Paired-end sequencing improves the mappability and specificity of read alignments, especially for large genomes.
Protocols
ChIP-exo 1.x
ChIP-exo 1.x improves on ChIP-seq by generating data with higher positional resolution by adding a lambda exonuclease digestion step. This higher resolution enables the capture of the organization of factors within a complex. Where version 1.0 was originally designed for the ABI SOLiD platform, ChIP-exo 1.1 makes the assay compatible with the Illumina NGS platform.56
ChIP-exo 2.x (ChIP-nexus)
ChIP-nexus utilizes a circular rather than linear DNA library, and increases efficiency of adapter ligation through CircLigase. However, the assay requires additional endonuclease digestion, and published ChIP-nexus data reports data loss due to poor barcode quality.7
ChIP-exo 3.x
ChIP-exo 3.x employs one-step adapter attachment using Tn5 tagmentation. This version of ChIP uses fewer steps than previous protocols while simultaneously retaining high resolution. However, the libraries produced may be enriched for longer fragments, since tagmentation by Tn5 may occur at a higher frequency for such fragments.6
ChIP-exo 4.x
ChIP-exo 4.x aims to streamline library construction and avoid the library biases of Tn5. ssDNA splint ligation is incorporated into the workflow. ChIP-exo 4.x is the simplest ChIP-exo version, but 4.0 may produce some steric exclusion of the adapter,and 4.1 may have lower precision.6
ChIP-exo 5.0
ChIP-exo 5.0 was developed to improve precision by reducing the "shouldering" found in versions 3.x and 4.x. Enzymatic steps are largely reduced, and as a result, library yield is greatly increased and signal concentration is maximized. 5.0 offers what the authors considered the best compromise in achieving high precision with a streamlined protocol at the time of publication.6
Advantages
- High resolution: ChIP-exo has been shown to give up to single base pair resolution in identifying protein binding locations. This is in contrast to ChIP-seq which can locate a protein's binding site only to with ±300 base pairs.4
- Lower rate of false positives: Contamination of non-protein-bound DNA fragments can result in a high rate of false positives and negatives in ChIP experiments. The addition of exonucleases to the process not only improves resolution of binding-site calling, but removes contaminating DNA from the solution before sequencing.4
- Proteins that are inefficiently bound to a nucleotide fragment are more likely to be detected by ChIP-exo. This has allowed, for example, the recognition of more CTCF transcription factor binding sites than previously discovered.5
- Lower sequencing requirements: Due to the higher resolution and reduced background, less depth of sequencing coverage is needed when using ChIP-exo.4
- Protein complex/Co-factor information: The direct crosslinking profiles from ChIP-exo data (primary peaks) can sometimes provide information about where on the DNA proteins interact. ChIP-exo data sometimes also captures positions of indirect crosslinking sites for secondary proteins ("piggybacking") in complex with the ChIP target (secondary peaks). These profiles can provide clues to the interaction between protein partners.8
Limitations
- Antibodies: As with any ChIP-based method, a suitable antibody for the protein of interest needs to be available in order to use this technique. Thus, the specificity, availability, and reproducibility of antibodies must be taken into consideration, or strains with epitope tags must be engineered.9
- Crosslinking: ChIP-exo uses formaldehyde crosslinking which has raised a variety of concerns within the genomics field, including the fact that cross-linking efficiency varies widely between different proteins.10 Certain targets may be "invisible" to ChIP-based approaches.
- Inaccessible heterochromatin: In part due to the crosslinking, more densely packed regions of the genome like heterochromatin are less extractable.11 As a result, it can be difficult to observe evidence of target binding in such regions using ChIP-based approaches.
- If a protein-DNA complex has multiple locations of cross-linking within a single binding event, then it can appear as though there are multiple distinct binding events. This likely results from these proteins being denatured and cross-linking at one of the available binding sites within the same event. The exonuclease would then stop at one of the bound sites, depending on which site the protein is cross-linked to.5 To get around this, there are certain peak calling methods (e.g. ChExMix) that take into account local crosslinking profiles to identify these multi-crosslink profiles as a single binding site.
Applications
Rhee and Pugh introduce ChIP-exo by performing analyses on a small collection of transcription factors: Reb1, Gal4, Phd1, Rap1 in yeast and CTCF in human. Reb1 sites were often found in clusters and these clusters had ~10-fold higher occupancy than expected. Secondary sites in clusters were found ~40 bp from a primary binding site. Binding motifs of Gal4 showed a strong preference for three of the four nucleotides, suggesting a negative interaction between Gal4 and the excluded nucleotide. Phd1 recognizes three different motifs which explains previous reports of the ambiguity of Phd1's binding motif. Rap1 was found to recognize four motifs.
Ribosomal protein genes bound by this protein had a tendency to use a particular motif with a stronger consensus sequence. Other genes often used clusters of weaker consensus motifs, possibly to achieve a similar occupancy. Binding motifs of CTCF employed four "modules". Half of the bound CTCF sites used modules 1 and 2, while the rest used some combination of the four. It is believed that CTCF uses its zinc fingers to recognize different combinations of these modules.5
Rhee and Pugh analyzed pre-initiation complex (PIC) structure and organization in Saccharomyces genomes. Using ChIP-exo, they were able to, among other discoveries, precisely identify TATA-like features in promoters reported to be TATA-less.12
Similar Methods
PB-exo
PB-exo was developed as an in vitro version of ChIP-exo (or "-exo" version of PB-seq13). Purified and sonicated naked genomic DNA is incubated with purified factors and then formaldehyde cross-linked. After this, the standard ChIP-exo protocol is followed.14 Like PB-seq, PB-exo provides information about genomic factor binding in the absence of chromatin structure or other secondary factor binding partners.
WhIP-exo
WhIP-exo is related to PB-exo except instead of or in addition to a purified target protein, the naked genomic DNA is incubated with crude whole-cell extract.14 This assays the genomic binding of a protein target in the presence of any potential cofactors. The set of available cofactors in the whole cell extract can be further curated through the modification of the source's genetic background.
PIP-seq
PIP-seq is a single-nucleotide resolution assay that determines the position of single-stranded DNA bound protein targets by combining ChIP-seq, ChIP-exo, and permanganate (KMNO4) footprinting techniques.1516 Permanganate treatment oxidizes single-stranded thymines, and after the target protein is immunoprecipitated out with the crosslinked DNA, piperidine treatment cleaves the DNA fragments at the oxidized thymines. Then, the library is prepared and sequenced. Bioinformatic filtering of reads for which the immediate upstream reference nucleotide is thymine ("T") enriches the signal for single-strand bound fragments and then downstream analysis can be performed.
See also
See also
References
References
- Gilmour, DS; JT Lis (1983). "Detecting protein-DNA interactions in vivo: Distribution of RNA polymerase on specific bacterial genes". Proceedings of the National Academy of Sciences. 81 (14): 4275–4279. Bibcode:1984PNAS...81.4275G. doi:10.1073/pnas.81.14.4275. PMC 345570. PMID 6379641.
- Albert, I; TN Mavrich; LP Tomsho; J Qi; SJ Zanton; SC Schuster; BF Pugh (2007). "Translational and rotational settings of H2A.Z nucleosomes cross the Saccharomyces cerevisiae genome". Nature. 446 (7135): 572–576. Bibcode:2007Natur.446..572A. doi:10.1038/nature05632. PMID 17392789. S2CID 4416890.
- Ren, B; F Robert; JJ Wyrick; O Aparicio; EG Jennings; I Simon; J Zeitlinger; J Schreiber; N Hannett; E Kan; et al. (2000). "Genome-wide location and function of DNA binding proteins". Science. 290 (5500): 2306–2309. Bibcode:2000Sci...290.2306R. CiteSeerX 10.1.1.123.6772. doi:10.1126/science.290.5500.2306. PMID 11125145.
- Pugh, Benjamin. "Methods, Systems and Kits for Detecting Protein-Nucleic Acid Interactions". United States Application Publication. United States Patents. Retrieved 17 February 2012.
- Rhee, Ho Sung; BJ Pugh (2011). "Comprehensive Genome-wide Protein-DNA Interactions Detected at Single-Nucleotide Resolution". Cell. 147 (6): 1408–1419. doi:10.1016/j.cell.2011.11.013. PMC 3243364. PMID 22153082.
- Rossi, Matthew; William KM Lai; B Franklin Pugh (2018). "Simplified ChIP-exo assays". Nat Commun. 9 (1): 2842. Bibcode:2018NatCo...9.2842R. doi:10.1038/s41467-018-05265-7. PMC 6054642. PMID 30030442.
- He, Qiye; Jeff Johnston; Julia Zeitlinger (2015). "ChIP-nexus enables improved detection of in vivo transcription factor binding footprints". Nat Biotechnol. 33 (4): 395–401. doi:10.1038/nbt.3121. PMC 4390430. PMID 25751057.
- Yamada, Naomi; Matthew J Rossi; Nina Farrel; B Franklin Pugh; Shaun Mahony (2020). "Alignment and quantification of ChIP-exo crosslinking patterns reveal the spatial organization of protein-DNA complexes". Nucleic Acids Res. 48 (20): 11215–11226. doi:10.1093/nar/gkaa618. PMC 7672471. PMID 32747934.
- Lai, William KM; Luca Mariani; Gerson Rothschild; Edwin R Smith; Bryan J Venters; Thomas R Blanda; Prashant K Kuntala; Kylie Bocklund; Joshua Mairose; Sarah N Dweikat; Katelyn Mistretta; Matthew J Rossi; Daniela James; James T Anderson; Sabrina K Phanor; Wanwei Zhang; Zibo Zhao; Avani P Shah; Katherine Novitzky; Eileen McAnarney; Michael-C Keogh; Ali Shilatifard; Uttiya Basu; Martha L Bulyk; B Franklin Pugh (2021). "A ChIP-exo screen of 887 Protein Capture Reagents Program transcription factor antibodies in human cells". Genome Res. 31 (9): 1663–1679. doi:10.1101/gr.275472.121. PMC 8415381. PMID 34426512.
- Gavrilov, Alexey; Sergey V Razin; Giacomo Cavalli (2015). "In vivo formaldehyde cross-linking: it is time for black box analysis". Brief Funct Genomics. 14 (2): 163–5. doi:10.1093/bfgp/elu037. PMC 6090872. PMID 25241225.
- Mansisidor, Andrés R; Viviana I Risca (2022). "Chromatin accessibility: methods, mechanisms, and biological insights". Nucleus. 13 (1): 236–276. doi:10.1080/19491034.2022.2143106. PMC 9683059. PMID 36404679.
- Rhee, Ho Sung; BJ Pugh (2012). "Genome-wide structure and organization of eukaryotic pre-initiation complexes". Nature. 483 (7389): 295–301. Bibcode:2012Natur.483..295R. doi:10.1038/nature10799. PMC 3306527. PMID 22258509.
- Guertin, Michael J; André L Martins; Adam Siepel; John T Lis (2012). "Accurate prediction of inducible transcription factor binding intensities in vivo". PLOS Genet. 8 (3) e1002610. doi:10.1371/journal.pgen.1002610. PMC 3315474. PMID 22479205.
- Rossi, Matthew J; William KM Lai; B Franklin Pugh (2018). "Genome-wide determinants of sequence-specific DNA binding of general regulatory factors". Nat Biotechnol. 28 (4): 497–508. doi:10.1101/gr.229518.117. PMC 5880240. PMID 29563167.
- Li, Jian; Yingyun Liu; Ho Sung Rhee; Saikat Kumar B Ghosh; Lu Bai; B Franklin Pugh; David S Gilmour (2013). "Kinetic competition between elongation rate and binding of NELF controls promoter-proximal pausing". Mol Cell. 50 (5): 711–22. doi:10.1016/j.molcel.2013.05.016. PMC 3695833. PMID 23746353.
- Lai, William KM; B Franklin Pugh (2017). "Genome-wide uniformity of human 'open' pre-initiation complexes". Genome Research. 27 (1): 15–26. doi:10.1101/gr.210955.116. PMC 5204339. PMID 27927716.
External links
External links
- DNA-protein interactions in high definition
- Resolving transcription factor binding
- High-resolution chromatin immunoprecipitation
- Important Gene-Regulation Proteins Pinpointed by New Method
- CexoR: An R/Bioconductor Package to Uncover High-resolution Protein-DNA Interactions in ChIP-exo Replicates
- Peconic Genomics